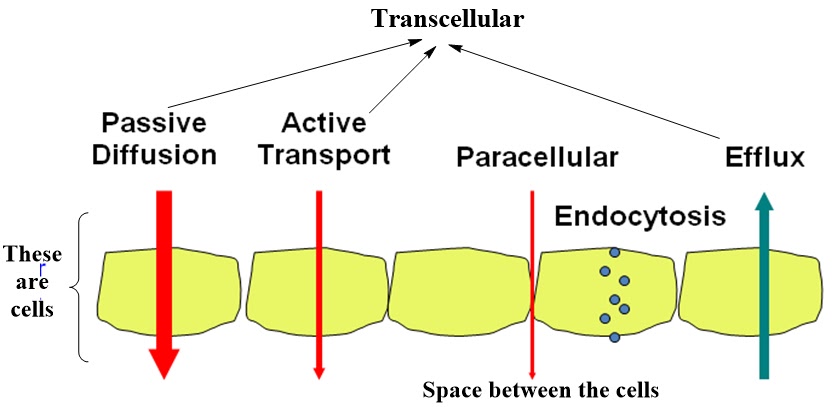
Will the test compound / drug compound remain circulating in
plasma within the body?
As mentioned earlier the liver is the major drug metabolizing
organ for the large majority of pharmaceutical drugs. For this reason, the in vitro models used to
investigate drug metabolism often focus on hepatocytes or subcellular fractions
of the liver such as microsomes, cytosol, S9 or mitochondria where
concentrations of particular enzymes are high.
Microsomes contain Phase I oxidative enzymes
including CYP enzymes.
Liver microsomes are subcellular fractions which
contain membrane bound drug metabolizing enzymes. These do not have an intact cell membrane.
The assay uses subcellular fractions of liver, microsomes, to investigate the metabolic fate of compounds. Liver microsomes consist mainly of endoplasmatic reticulum and contain many drug-metabolizing enzymes, including cytochrome P450s (CYPs), flavin monooxygenases, carboxylesterases, and epoxide hydrolase.
The assay uses subcellular fractions of liver, microsomes, to investigate the metabolic fate of compounds. Liver microsomes consist mainly of endoplasmatic reticulum and contain many drug-metabolizing enzymes, including cytochrome P450s (CYPs), flavin monooxygenases, carboxylesterases, and epoxide hydrolase.
Liver microsomes are available
commercially as frozen preparations that are usually prepared in bulk with
pooled livers from sacrificed mice, rat or human cadavers (corpse/dead bodies).
Addition of relevant co-factor to the incubation is
necessary (NADPH and UDPGA are added.
These are cofactors which are needed to initiate the Phase I metabolic
reactions)
The
use of species-specific microsomes can be used to enable an understanding of
interspecies differences. [for example along with HLM (Human liver microsomes)
– MLM (mouse liver microsomes) and RLM (rat liver microsomes) are used]
___________
Most often, metabolic stability of compounds are assessed at a single
concentration (typically 10 μM) at t = 0 and at t = 60 min. Stability of
compounds are tested in human (other species available) liver microsomes.
___________
S9 fraction
is
the post-mitochondrial supernatant fraction.
It contains both cytosolic and microsomal enzymes. This is to identify if cytosolic enzymes are
responsible for the formation of a metabolite.
The advantage of using S9 fraction for in vitro screening is that it contains a
wide variety of both phase I and phase II enzymes.
Addition of relevant co-factor to the incubation is
necessary [cofactors such as UDPGA and PAPS to investigate Phase II metabolic
pathways.]
Hepatocytes are more representative of the in vivo situation because they contain a
cell membrane and do not require additional co-factors. Hepatocytes contain full complement of
enzymes for both Phase I and Phase II metabolism.
The use of species-specific cryopreserved
hepatocytes can be used to enable an understanding of interspecies differences
in metabolism.
CYP inhibition studies:
·
CYPs constitute a superfamily of heme
enzymes
·
CYPs play a major role in the metabolism
of a wide array of xenobiotics including drugs, chemical carcinogens,
insecticides, petroleum products, and other environmental pollutants.
·
Although the liver is the primary organ
for drug metabolism, extrahepatic tissues such as lung, kidney and intestine,
also play an important role for detoxification or biotransformation of
xenobiotics. Each tissue has a unique P450 isozyme distribution and regulatory
mechanism for cytochrome P450 gene (CYP) expression.
v In vitro
cytochrome P450 inhibition data are useful in designing strategies for
investigating clinical Drug-Drug interaction Studies
v Assessment
of the potential of a compound to inhibit a specific cytochrome P450 enzyme is
important as co-administration of compounds may result in one or both
inhibiting the other’s metabolism. This may affect plasma levels in vivo and
potentially lead to adverse drug reactions or toxicity
v CYP1A2,
CYP2C8, CYP2C9, CYP2C19, CYP2D6, CYP3A4 are few important CYP450 enzymes. Identifying which of these enzymes are
responsible for drug metabolism is important.
v All
of these cytochrome P450 inhibition reactions are
incubated separately for each isoform.
v CYP450
3A4 is of paramount importance, because it is the most abundant P450 in the
human liver and is known to metabolize the majority of drugs whose
biotransformation is known.